
La substitució genètica que pot canviar el destí de l'Oriol: “Una petita millora seria al·lucinant”

LLEGIR EN CASTELLÀ
L’Oriol no parla. No camina ni manipula; no agafa ni assenyala. Res de tot això formava part del món al qual ell podia accedir mentre creixia, ni tampoc ho podien imaginar els seus pares, els mallorquins David Campaner i Queta Socías, quan el van tenir per primera vegada als braços. Amb el pas del temps, descobririen que el seu fill habitava un món sense paraules, sense gestos funcionals i sense moviments voluntaris reconeixibles. Un món que, lluny de ser buit, ells han après a llegir, a interpretar i, sobretot, a compartir amb ell.
L’entrevista té lloc en un d’aquells moments que formen part de la rutina familiar: són les 20 hores i a l’Oriol li toca anar-se’n al llit. Mentrestant, el seu pare, David, parla amb serenor, amb aquell to que només s’adquireix quan la vida t’obliga a conviure durant anys amb allò extraordinari. Ho fa sobre el seu fill Oriol. Avui té 14 anys i és l’únic cas diagnosticat a les Balears de la síndrome FOXG1, una malaltia neurològica rara, genètica, devastadora… però, potser per primera vegada, amb una sortida real a la vista.
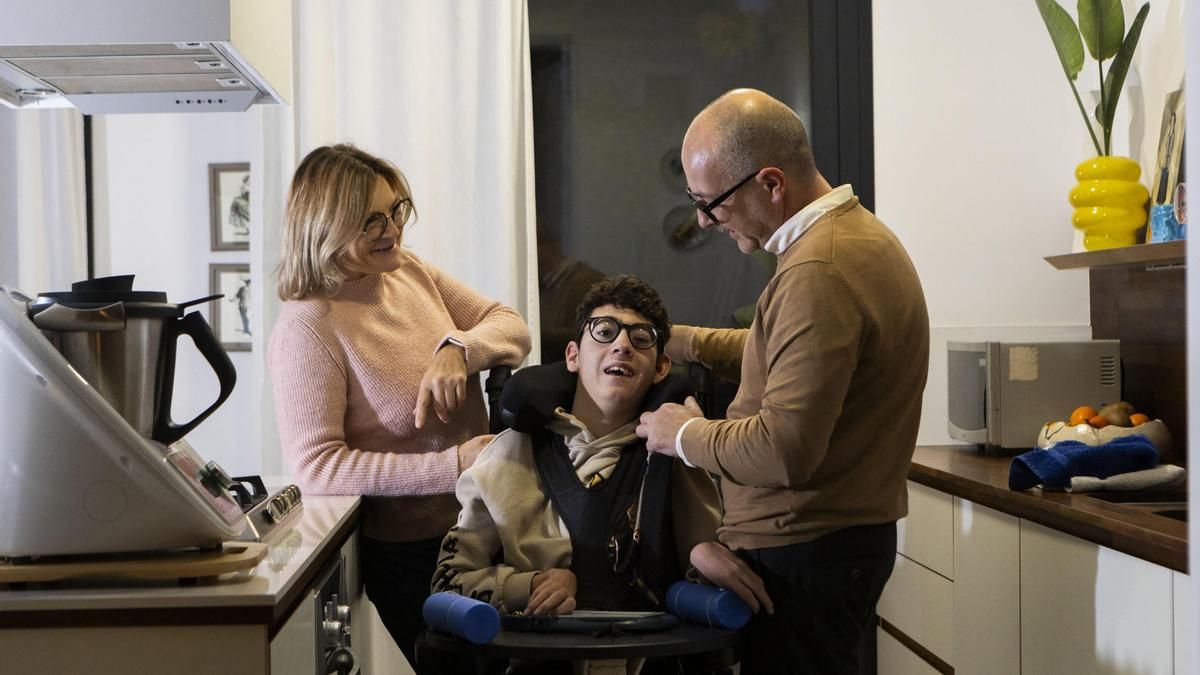

El dia a dia del jove és el d’una persona amb dependència absoluta. “Ell no parla, no camina, no manipula, no agafa… cal fer-li-ho tot”, explica el seu pare sense dramatisme, amb la naturalitat de qui ha normalitzat allò que mai no hauria d’haver estat normal. No existeix una comunicació convencional i qualsevol persona que no hi convisqui, reconeix David, “no notaria cap mena de comunicació significativa”. Tanmateix, hi ha matisos. L’Oriol té una mica de contacte visual. Mira. De vegades costa, de vegades cal esperar, però ho aconsegueix. I no mira tothom de la mateixa manera. És selectiu: a algunes persones les observa i somriu; a d’altres, no. “La mirada és una evolució enorme. Fa uns anys aquest estadi no existia”, subratlla. En el cas de l’Oriol, cada petit avanç és un esdeveniment.
'Ell no parla, no camina, no manipula, no agafa… cal fer-li-ho tot', explica el seu pare. L'Oriol utilitza la mirada per comunicar-se. 'És una revolució enorme', afegeix el progenitor
Tot això ja hi era des del naixement, però en tractar-se d’una malaltia genètica, ningú no la va poder detectar d’immediat. No va ser fins que l’Oriol va complir dos anys que alguna cosa va començar a no quadrar. La parla no arribava, els balboteigs no es materialitzaven i la seva evolució no s’assemblava a la d’altres infants de la seva edat. Aquí va començar una autèntica odissea familiar. Primer van passar per hospitals privats de Mallorca, després per la sanitat pública, amb Son Espases com a primer punt de referència. Però les respostes no arribaven. El diagnòstic definitiu va aparèixer fora de l’illa, a l’Hospital Sant Joan de Déu de Barcelona, després de viatges constants cada dues o tres setmanes, proves interminables i una espera emocionalment demolidora. Allà van posar nom al que li passava a l’Oriol: síndrome FOXG1.

Nombre de casos
A Espanya hi ha 36 casos registrats d’aquesta malaltia rara. Existeix una associació nacional, sense seu física, però estretament vinculada a la fundació internacional que impulsa la investigació des dels Estats Units, la FOXG1 Research Foundation. “El primer dia a Sant Joan de Déu no l’oblidarem mai”, recorda David. “Va ser un dia en què ens vam oblidar que teníem un fill amb discapacitat. Ens van tractar com a pares i l’Oriol com un nen. Allà va començar una relació preciosa amb els sanitaris”. Ho diu amb convicció: “Sí que es pot aconseguir que el teu fill sigui atès com el teu fill i no com algú amb discapacitat”.
El primer dia a l'Hospital Sant Joan de Déu no l'oblidarem mai. Va ser un dia en què se'ns va oblidar que teníem un fill discapacitat. Ens van tractar com a pares i a l'Oriol com a un nen
La síndrome FOXG1 és un trastorn neurològic rar causat per una mutació en el gen FOXG1, un dels gens clau en el desenvolupament cerebral durant l’etapa embrionària. Conegut també com a Brain factor 1, aquest gen activa molts altres gens fonamentals per al cervell. La majoria dels infants afectats no parlen, presenten discapacitats físiques i cognitives greus, epilèpsia intratable, trastorns del moviment, dificultats respiratòries, problemes de visió i d’alimentació. Es calcula que hi ha uns 1.500 casos a tot el món, tot i que el nombre de diagnòstics augmenta any rere any. La mutació sol ser de novo, és a dir, espontània i no heretada. La seva investigació podria ajudar a trobar solucions per a altres malalties com l’Alzheimer, l’esquizofrènia o l’epilèpsia.
L’Oriol va començar a ser estudiat a Barcelona per la doctora en genètica Judith Armstrong, on es van recollir cultius cel·lulars per investigar el seu cas. Aquella línia de treball va fer un gir quan la Universitat de Buffalo, als Estats Units, va assumir el lideratge de la investigació. I és que aquest campus és avui l’epicentre mundial de l’estudi del FOXG1. Des de Sant Joan de Déu es van compartir totes les dades clíniques de l’Oriol amb l’equip nord-americà, vinculat a la FOXG1 Research Foundation, una entitat que, com tantes vegades passa en les malalties rares, va néixer de l’empenta d’uns pares que es van negar a rendir-se, ja que el seu fill pateix la mateixa síndrome.

Substitució genètica
“Posa la pell de gallina”, diu David en parlar del punt en què es troba avui la investigació. Perquè allò que s’està desenvolupant podria arribar a ser una cura (el tractament no desfaria els danys cerebrals provocats durant el desenvolupament), tot i que resulti gairebé impossible d’imaginar quan s’observa l’estat actual de l’Oriol. La via és una teràpia de substitució genètica, una tècnica absolutament innovadora, combinada amb intel·ligència artificial, que ja ha estat provada amb èxit en ratolins. Tal com explica a elDiario.es el genetista Lluís Montoliu, investigador científic del CSIC i sotsdirector del Centre Nacional de Biotecnologia (CNB), als Estats Units uns investigadors han preparat un model animal de la síndrome amb rosegadors mitjançant la CRISPR (una tècnica d’edició genètica revolucionària, ja que es basa en el descobriment que les proteïnes Cas tallen l’ADN sempre que se’ls proporcioni un ARN de reconeixement adequat). Com que l’ARN es pot sintetitzar al laboratori, les possibilitats podrien ser infinites.
La Universitat de Buffalo, als Estats Units, estudia una teràpia de substitució genètica, combinada amb intel·ligència artificial, que ja ha estat provada amb èxit en rates


La FOXG1 és una mutació cel·lular que impedeix que determinades cèl·lules del sistema nerviós facin la seva funció. La teràpia consisteix a introduir una nova codificació genètica mitjançant una simple injecció. Un vector viral transporta el FOXG1 sa fins a les cèl·lules necessàries, substitueix la proteïna defectuosa i ajuda les cèl·lules a funcionar amb normalitat. “Els estudis estarien lluny, de moment, de convertir-se en una aplicació clínica, en una teràpia que es pugui oferir des dels hospitals”, objecta el genetista, que tanmateix explica que la recerca i els resultats en el model animal “són prometedors”, tot i saber que hi ha limitacions (rosegadors i humans comparteixen alguns paral·lelismes fisiològics, metabòlics i anatòmics, encara que no tots).
Afegeix que les teràpies van encaminades a aturar el deteriorament neuronal progressiu i irreversible provocat per aquesta malaltia i que, en alguns rosegadors —que, a diferència dels humans, tenen visió nocturna—, s’ha vist que alguns dels símptomes que presentava el model animal no desapareixen, però sí que milloren. Amb tot, el 2026 començaran els assaigs clínics en humans, tant a Buffalo com en altres punts dels Estats Units i d’Europa.
Per arribar fins aquí, encara és imprescindible recaptar fons. La família i l’associació impulsen mercats solidaris, venda de samarretes, donacions particulars i la implicació d’empresaris que desitgen formar part d’un avenç que podria marcar un abans i un després. Cal l’última gran partida econòmica. I hi ha una dada clau: aquesta investigació no només beneficiarà el FOXG1. És una malaltia ‘germana’ de la síndrome de Rett i la solució genètica que s’està desenvolupant podria obrir portes a moltes altres patologies rares. No és una quimera: ja ha passat abans en altres disciplines. El 2025, onze infants amb sordesa incurable van tornar a sentir gràcies a la teràpia gènica.
Aquesta investigació pionera també podria beneficiar altres patologies rares, com la síndrome de Rett
Treballar l’estimulació
Quan se li pregunta per les rutines del seu fill, David recorre a l’humor per explicar: “És com qualsevol altre adolescent de 14 anys… una marmota que no es desperta”. L’Oriol estudia a l’Associació de Paràlisi Cerebral i Malalties Afins (ASPACE), en una aula amb quatre alumnes més amb altres discapacitats. Allà treballen l’estimulació, la comunicació mitjançant el sistema Tobii —lectura de pupil·les i plafons visuals—. Rep tractaments de fisioteràpia i treballa amb música, veu pel·lícules i també hi ha temps perquè pugui estar amb els amics tranquilament. “Cal deixar tanta canya”, diu el seu pare mentre somriu.
El FOXG1 no entén d’estàndards. Hi ha infants que caminen, d’altres que es comuniquen. Aquest no és el cas de l’Oriol, que a més pateix epilèpsia refractària, amb manifestacions molt diferents de les habituals. En el seu cas, una crisi pot aparèixer com a moviments laterals de la mandíbula, dilatació de pupil·les, caigudes sobtades del cap i ulls cap amunt o fins i tot una rialla histriònica. “Ha tingut molts patrons diferents. No ens cansem… podem fer una conga amb tot”, fa broma David.

“Amb l’Oriol cal tenir una empresa”, resumeix el pare. Ell i la Queta es reparteixen funcions, torns i responsabilitats, atents les 24 hores del dia. No es pot abaixar la guàrdia. El vincle més clar de l’Oriol amb l’art és la música. Li encanta l’últim disc de Rosalía, el fado portuguès, algunes peces de Maria Callas —no totes— i una mica de reggaeton que escolta a l’escola. Al nucli domèstic toca, a la seva manera, un piano Casio. De petit, quan anava amb el seu exoesquelet, ho tenia clar: sempre s’abalançava sobre les tecles.
A l'Oriol li agrada l'últim disc de Rosalía, el fado portuguès, algunes peces de Maria Callas —no totes— i una mica de reggaeton que escolta a l'escola
El que cobreix el sistema públic
Les institucions cobreixen els vols i les dietes per a les revisions a Barcelona i part del material ortopèdic, però les mancances són notables. A tall d’exemple, un bipedestador costa uns 7.000 euros i avui l’ajuda tot just cobreix el 30%. El mateix passa amb un caminador. L’Oriol té escoliosi i les aportacions públiques resulten insuficients per a la família. Actualment camina amb un NF-Walker, un estabilitzador que la família va aconseguir a Bilbao després d’una recerca gairebé artesanal per ortopèdies de tot el país.
Amb tot, David llança una petició clara: empatia i escolta. “Els pares no som metges però sí que vivim el dia a dia. En les malalties rares no hi ha patrons tancats”. I mira cap al futur amb una barreja de prudència i esperança: “Ens crida molt l’atenció pensar amb què ens podríem trobar amb l’Oriol si finalment es dona la modificació genètica. Si hi ha una petita millora… això ja seria al·lucinant”.

L’Oriol Campaner i els seus pares formen una família petita però sòlida. Cada tarda, quan el jove torna a casa, l’“etern nadó” necessita totes les atencions. Bolquer, cures constants i una entrega absoluta. I tot i així, la paraula que més es repeteix no és resignació, sinó esperança. Perquè potser, gràcies a la substitució genètica, el destí de l’Oriol i de centenars d’infants arreu del món està a punt de canviar. I perquè, una vegada més, tot va començar amb uns pares que es van negar a acceptar que no hi havia res a fer.

0