
La sustitución genética que puede cambiar el destino de Oriol: “Una pequeña mejoría sería alucinante”

LEER ESTE TEXTO EN CATALÁN
Oriol no habla. No camina ni manipula; no coge ni señala. Nada de eso estaba en el mundo que Oriol podía alcanzar al crecer, ni tampoco podían imaginarlo sus padres, los mallorquines David Campaner y Queta Socías, cuando lo tuvieron por primera vez entre los brazos. Con el paso del tiempo, descubrirían que su hijo habitaba un mundo sin palabras, sin gestos funcionales y sin movimientos voluntarios reconocibles. Un mundo que, lejos de ser vacío, ellos han aprendido a leer, a interpretar y, sobre todo, a compartir con él.
La entrevista se produce en uno de esos momentos que forman parte de la rutina familiar: son las 20 horas y a Oriol le toca meterse en la cama. Mientras, su padre, David, habla con serenidad, con ese tono que solo se adquiere cuando la vida te obliga a convivir durante años con lo extraordinario. Lo hace sobre su hijo Oriol. Tiene hoy 14 años y es el único caso diagnosticado en Balears del síndrome FOXG1, una enfermedad neurológica rara, genética, devastadora… pero, quizá por primera vez, con una salida real a la vista.
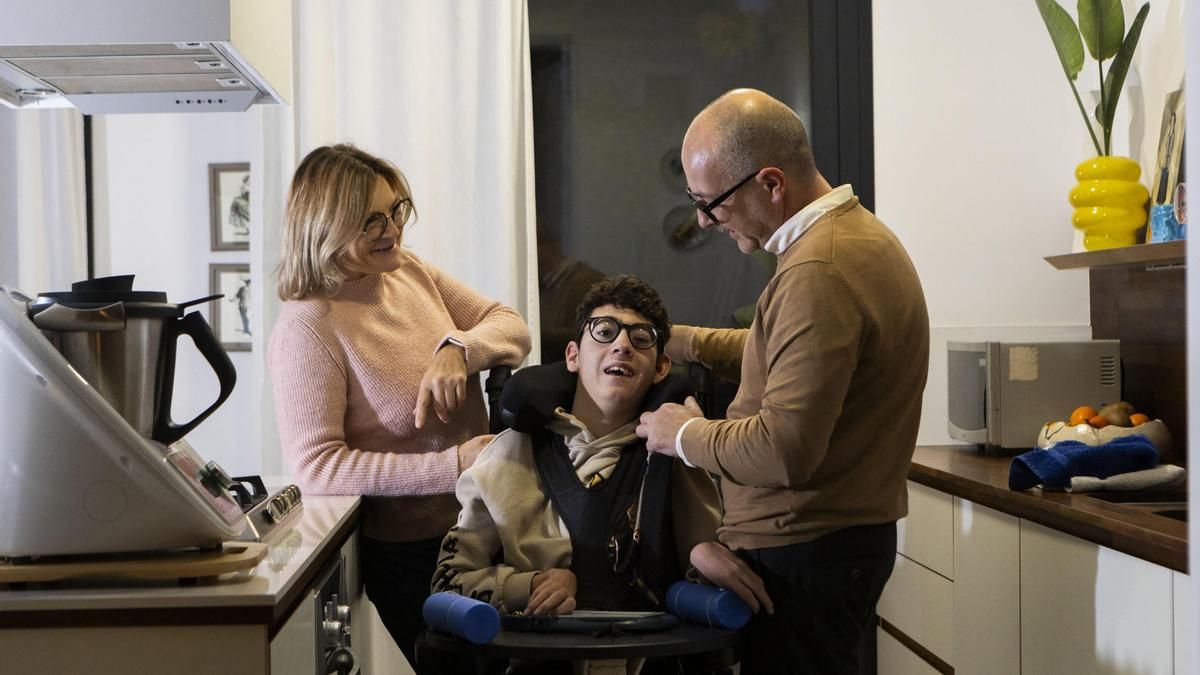

El día a día de Oriol es el de una persona con dependencia absoluta. “Él no habla, no camina, no manipula, no coge… hay que hacérselo todo”, explica su padre sin dramatismo, con la naturalidad de quien ha normalizado lo que nunca debió ser normal. No existe una comunicación convencional y cualquier persona que no conviva con él, reconoce David, “no notaría ningún tipo de comunicación significativa”. Sin embargo, hay matices. Oriol tiene algo de contacto visual. Mira. A veces cuesta, a veces hay que esperar, pero lo consigue. Y no mira a todo el mundo por igual. Es selectivo: a algunas personas las observa y sonríe; a otras, no. “La mirada es una evolución enorme. Hace unos años ese estadio no existía”, subraya. En el caso de Oriol, cada pequeño avance es un acontecimiento.
'Él no habla, no camina, no manipula, no coge… hay que hacérselo todo', explica su padre. Oriol utiliza la mirada para comunicarse. 'Es una revolución enorme', añade el progenitor
Todo esto estaba ya ahí desde el nacimiento, pero, al tratarse de una enfermedad genética, nadie pudo detectarla de inmediato. No fue hasta que Oriol cumplió dos años cuando algo empezó a no cuadrar. El habla no llegaba, los balbuceos no se materializaban y su evolución no se parecía a la de otros niños de su edad. Ahí comenzó una auténtica odisea familiar. Primero pasaron por hospitales privados de Mallorca, después por la sanidad pública, con Son Espases como punto de referencia inicial. Pero las respuestas no llegaban. El diagnóstico definitivo apareció fuera de la isla, en el Hospital Sant Joan de Déu de Barcelona, tras viajes constantes cada dos o tres semanas, pruebas interminables y una espera emocionalmente demoledora. Allí pusieron nombre a lo que le ocurría a Oriol: síndrome FOXG1.

Número de casos
En España hay 36 casos registrados de esta enfermedad rara. Existe una asociación nacional, sin sede física, pero estrechamente vinculada a la fundación internacional que impulsa la investigación desde Estados Unidos, la FOXG1 Research Foundation.
“El primer día en Sant Joan de Déu no lo olvidaremos nunca”, recuerda David. “Fue un día en el que se nos olvidó que teníamos un hijo discapacitado. Nos trataron como padres y a Oriol como a un niño. Ahí empezó una relación preciosa con los sanitarios”. Lo dice con convicción: “Sí que se puede conseguir que a tu hijo lo atiendan como a tu hijo y no como a alguien discapacitado”.
El primer día en Hospital Sant Joan de Déu no lo olvidaremos nunca. Fue un día en el que se nos olvidó que teníamos un hijo discapacitado. Nos trataron como padres y a Oriol como a un niño
El síndrome FOXG1 es un trastorno neurológico raro causado por una mutación en el gen FOXG1, uno de los genes clave en el desarrollo cerebral durante la etapa embrionaria. Conocido también como Brain factor 1, este gen activa muchos otros genes fundamentales para el cerebro. La mayoría de los niños afectados no hablan, presentan discapacidades físicas y cognitivas graves, epilepsia intratable, trastornos del movimiento, dificultades respiratorias, problemas de visión y de alimentación. Se calcula que hay unos 1.500 casos en todo el mundo, aunque el número de diagnósticos aumenta año tras año. La mutación suele ser de novo, es decir, espontánea y no heredada. Su investigación podría ayudar en la solución de otras enfermedades como el Alzheimer, la esquizofrenia o la epilepsia.
Oriol comenzó a ser estudiado en Barcelona por la doctora en genética Judith Armstrong, donde se recogieron cultivos celulares para investigar su caso. Aquella línea de trabajo dio un giro cuando la Universidad de Buffalo, en Estados Unidos, asumió el liderazgo de la investigación. Y es que ese campus es hoy el epicentro mundial del estudio del FOXG1. Desde Sant Joan de Déu se compartieron todos los datos clínicos de Oriol con el equipo estadounidense, vinculado a la FOXG1 Research Foundation, una entidad que, como tantas veces ocurre en las enfermedades raras, nació del empuje de unos padres que se negaron a rendirse, ya que su hijo sufre el mismo síndrome.

Sustitución genética
“Pone la piel de gallina”, dice David al hablar del punto en el que se encuentra hoy la investigación. Porque lo que se está desarrollando podría llegar a ser una cura (el tratamiento no desharía los daños cerebrales provocados durante el desarrollo), aunque resulte casi imposible de imaginar cuando se observa el estado actual de Oriol. La vía es una terapia de sustitución genética, una técnica absolutamente innovadora, combinada con inteligencia artificial, que ya ha sido probada con éxito en ratones. Como explica a elDiario.es el genetista Lluís Montoliu, investigador científico del CSIC y vicedirector del Centro Nacional de Biotecnología (CNB), en EEUU unos investigadores han preparado un modelo animal del síndrome con roedores mediante la CRISPR (técnica de edición genética revolucionaria, ya que se basa en el descubrimiento de que las proteínas Cas cortan el ADN siempre que se les proporcione un ARN de reconocimiento adecuado). Como el ARN se puede sintetizar en el laboratorio, las posibilidades podrían ser infinitas.
La Universidad de Buffalo, en Estados Unidos, estudia una terapia de sustitución genética, combinada con inteligencia artificial, que ya ha sido probada con éxito en ratas


La FOXG1 es una mutación celular que impide que determinadas células del sistema nervioso hagan su función. La terapia consiste en introducir una nueva codificación genética mediante una simple inyección. Un vector viral transporta el FOXG1 sano hasta las células necesarias, reemplaza la proteína defectuosa y ayuda a las células a funcionar con normalidad. “Los estudios estarían lejos por el momento de convertirse en una aplicación clínica, en una terapia que se pueda ofertar desde los hospitales”, objeta el genetista, quien, sin embargo, cuenta que la investigación y los resultados en el modelo animal “son prometedores”, aun sabiendo que hay limitaciones (comparten los roedores y los humanos algunos paralelismos fisiológicos, metabólicos y anatómicos, aunque no todos).
Añade que las terapias van encaminadas a detener el deterioro neuronal progresivo e irreversible provocado por esta enfermedad y que en algunos roedores -que a diferencia de los humanos tienen visión nocturna- se ha visto que algunos de los síntomas que tenía el modelo animal no desaparecen, pero sí mejoran. Con todo, en 2026 comenzarán los ensayos clínicos en humanos, tanto en Buffalo como en otros puntos de Estados Unidos y Europa.
Para llegar hasta ahí, todavía es imprescindible recaudar fondos. La familia y la asociación impulsan mercadillos, venta de camisetas, donaciones particulares y la implicación de empresarios que desean formar parte de un avance que podría marcar un antes y un después. Se necesita la última gran partida económica. Y hay un dato clave: esta investigación no solo beneficiará al FOXG1. Es una enfermedad ‘hermana’ del síndrome de Rett y la solución genética que se está desarrollando podría abrir puertas a muchas otras patologías raras. No es una quimera: ya ha ocurrido antes en otras disciplinas. En 2025 once niños con sordera incurable volvieron a oír gracias a la terapia génica.
Esta investigación pionera también podría beneficiar a otras patologías raras, como el síndrome de Rett
Trabajar la estimulación
Cuando se le pregunta por las rutinas de Oriol, David recurre al humor para explicar: “Es como cualquier otro adolescente de 14 años… una marmota que no se despierta”. Oriol estudia en la Asociación de Parálisis Cerebral y Enfermedades Afines (ASPACE), en un aula con otros cuatro alumnos con otras discapacidades. Allí trabajan la estimulación, la comunicación mediante el sistema Tobii —lectura de pupilas y tableros visuales—. Recibe tratamientos con fisioterapia y trabajan con música, ven películas y también hay tiempo para que estén tranquilos los amigos. “Hay que dejar de tanto machaque”, dice su padre mientras sonríe.
El FOXG1 no entiende de estándares. Hay niños que caminan, otros que se comunican. Ese no es el caso de Oriol, quien además padece de epilepsia refractaria, con manifestaciones muy distintas a las habituales. En su caso, una crisis puede aparecer como movimientos laterales de mandíbula, dilatación de pupilas, caídas repentinas de cabeza y ojos hacia arriba o incluso una risa histriónica. “Ha tenido muchos patrones distintos. No nos cansamos… podemos hacer una conga con todo”, bromea David.

“Con Oriol hay que tener una empresa”, resume el padre. Él y Queta se reparten funciones, turnos y responsabilidades, atentos las 24 horas del día. No se puede bajar la guardia. El vínculo más claro de Oriol con el arte es la música. Le encanta el último disco de Rosalía, el fado portugués, algunas piezas de Maria Callas —no todas— y algo de reguetón que escucha en el colegio. En el núcleo doméstico toca, a su manera, un piano Casio. De pequeño, cuando iba con su exoesqueleto, lo tenía claro: siempre se abalanzaba sobre las teclas.
A Oriol le encanta el último disco de Rosalía, el fado portugués, algunas piezas de Maria Callas —no todas— y algo de reguetón que escucha en el colegio
Lo que cubre el sistema público
Las instituciones cubren los vuelos y dietas para las revisiones en Barcelona y parte del material ortopédico, pero las carencias son notables. A modo de ejemplo, un bipedestador –un sistema de verticalización– cuesta unos 7.000 euros y hoy la ayuda apenas cubre el 30%. Lo mismo ocurre con un caminador. Oriol tiene escoliosis y las aportaciones públicas resultan insuficientes para la familia. Actualmente, camina con un NF-Walker, un estabilizador que la familia consiguió en Bilbao tras una búsqueda casi artesanal por ortopedias de todo el país.
Con todo, David lanza una petición clara: empatía y escucha. “Los padres no somos médicos, pero sí vivimos el día a día. En las enfermedades raras no hay patrones cerrados”. Y mira al futuro con una mezcla de prudencia y esperanza: “Nos llama mucho la atención pensar con qué Oriol nos podríamos encontrar si finalmente se da la modificación genética. Si hay una pequeña mejoría… eso ya sería alucinante”.

Oriol Campaner y sus padres conforman una familia pequeña, pero sólida. Cada tarde, cuando el joven vuelve a casa, el “eterno bebé” necesita todas las atenciones. Pañal, cuidados constantes y una entrega absoluta. Y aun así, la palabra que más se repite no es resignación, sino esperanza. Porque quizá, gracias a la sustitución genética, el destino de Oriol y de cientos de niños en todo el mundo esté a punto de cambiar. Y porque, una vez más, todo empezó con unos padres que se negaron a aceptar que no había nada que hacer.

6